SARS-CoV-2-surveillance via het riool: praktisch en efficiënt
11-2-2022Tijdens een pandemie is virus-surveillance uiterst belangrijk: het monitoren van de verspreiding en evolutie van het virus. Daarbij gaat het niet alleen om het aantal besmettingen, maar ook om het type besmettingen: om welke variant van het virus gaat het? Een virus muteert continu, en sommige van deze mutanten hebben een voordeel ten opzichte van anderen, bijvoorbeeld omdat ze zich sneller verspreiden. Een variant wordt gedefinieerd door een combinatie van mutaties in het genoom van het virus. Zo hebben we inmiddels verschillende varianten SARS-CoV-2 gezien, elk weer besmettelijker dan de vorige: eerst Alpha, toen Delta en nu Omikron. Door de verspreiding van nieuwe varianten te monitoren kunnen gepaste maatregelen genomen worden.
In de afgelopen twee jaar is rioolwater een rijke bron van informatie rondom de verspreiding van het virus gebleken. Besmette personen scheiden virusdeeltjes uit via urine en ontlasting, en deze zijn terug te vinden in het rioolwater. Het virus-RNA kan uit het rioolwater gefilterd worden en vervolgens wordt de concentratie SARS-CoV-2 deeltjes gemeten; dit blijkt een goede voorspelling te geven van het aantal besmettingen in de samenleving. Terwijl niet iedereen die besmet is zich laat testen bij de GGD, bijvoorbeeld omdat sommige besmettingen asymptomatisch zijn, gaat iedereen wel naar het toilet. Het rioolwater kan dus een completer beeld geven van de hoeveelheid besmettingen op een bepaalde geografische locatie. Bovendien worden op deze manier via een enkel monster wel duizenden mensen tegelijk getest: surveillance via het riool is dus praktisch en efficiënt.
Echter, het is niet eenvoudig om onderscheid te maken tussen verschillende varianten van het virus op basis van rioolwateronderzoek. Met behulp van ‘genome sequencing’ kan weliswaar het genetisch materiaal van SARS-CoV-2 dat zich ophoopt in het rioolwater uitgelezen worden, maar dit is het genetisch materiaal van vele infecties samen in één dataset (figuur 1a). Het afleiden van de verhouding van verschillende varianten van het virus uit deze data is een wiskundig probleem dat extra lastig is door de onbetrouwbaarheid van de data: vaak is de concentratie virusdeeltjes laag waardoor de genomen incompleet zijn, en er kunnen tijdens het sequencen ook nog eens leesfouten gemaakt worden door de sequencingmachine.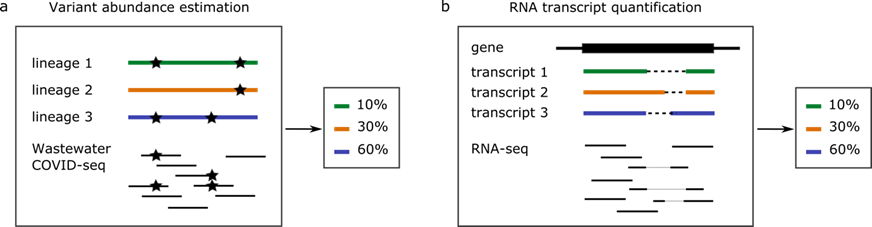
Figuur 1. Schematische weergave van twee vergelijkbare kwantificatie problemen op basis van sequencing data.
- a) Het kwantificeren van virus varianten met behulp van genetisch materiaal van het virus in rioolwatermonsters. Sterren geven hier mutaties aan ten opzichte van het originele virus. Lineage 1, 2 en 3 zijn verschillende varianten van het virus. Het doel van "variant abundance estimation" is om de relatieve hoeveelheid van elk van deze varianten te bepalen.
- b) Het kwantificeren van genexpressie in cellen met behulp van RNA-sequencing. De eerste stap van genexpressie is transcriptie: het overschrijven van het gen (DNA) naar mRNA. Dit kan op verschillende manieren, waarbij verschillende stukken uit het mRNA weggeknipt worden. Dit leidt tot transcripties 1, 2 en 3. Het doel van "RNA transcript quantification" is om de relatieve hoeveelheid van elk van de transcripties te bepalen.
Onderzoekers Jasmijn Baaijens (TU Delft) en Michael Baym (Harvard Medical School) hebben laten zien dat dit probleem, wiskundig gezien, vrijwel identiek is aan een ander probleem in de bioinformatica: het kwantificeren van genexpressie in cellen met behulp van RNA-sequencing (figuur 1b). Voor dit probleem zijn al vele algoritmes ontwikkeld, welke vrijwel direct toegepast kunnen worden voor het kwantificeren van SARS-CoV-2-varianten in het rioolwater. Een vereiste hiervoor is wel dat de genomen van de virusvarianten die we willen kwantificeren bekend zijn, deze moeten namelijk aangeleverd worden als ‘reference set’. De onderzoekers hebben daarom software geschreven die een zorgvuldige selectie maakt uit de SARS-CoV-2-database GISAID [1], passend bij de regio en tijdsbestek waarin de monsters genomen zijn. Vervolgens kan met kallisto [2], een veelgebruikte methode voor RNA-seq kwantificatie, de relatieve hoeveelheid van de verschillende virusvarianten geschat worden.
De onderzoeksresultaten [3] op gesimuleerde data wijzen uit dat, in theorie, de voorspelde frequenties in deze toepassing erg nauwkeurig zijn. Om de betrouwbaarheid in de praktijk te beoordelen hebben de onderzoekers gedurende vier maanden tijd (januari t/m april 2020) iedere twee dagen een rioolwatermonster genomen en geanalyseerd. Vervolgens hebben ze deze voorspellingen vergeleken met de frequenties afgeleid uit het steekproefsgewijs analyseren van individuele patiënten en de trends bleken goed overeen te komen. Met behulp van dit algoritme kan dus eenvoudig op grote schaal de verspreiding van verschillende varianten van SARS-CoV-2 gevolgd worden. Wel merken de onderzoekers op dat individuele monsters onbetrouwbaar zijn door een mogelijk tekort aan virusmateriaal en andere toevalsfactoren. Op dit moment is de methode enkel geschikt voor het kwantificeren van virusvarianten waarvan het genoom al bekend is; in de toekomst willen de onderzoekers de methode uitbreiden zodat ook het ontdekken van nieuwe mutanten mogelijk wordt.
Onderzoekers:
Dit onderzoek is het resultaat van een samenwerking tussen meerdere universiteiten [3]. De onderzoekers die hierin een hoofdrol hebben gehad, zijn:
1) Jasmijn Baaijens - Assistant professor of bioinformatics (j.a.baaijens@tudelft.nl)
Pattern Recognition & Bioinformatics, Department of Intelligent Systems, TU Delft
2) Alessandro Zulli - PhD student
Department of Chemical and Environmental Engineering, Yale University, USA
3) Isabel Ott - Lab assistant
Department of Epidemiology of Microbial Diseases, Yale School of Public Health, USA
4) Nathan Grubaugh - Associate professor of epidemiology
Department of Epidemiology of Microbial Diseases, Yale School of Public Health, USA
5) Jordan Peccia - Professor of environmental engineering
Department of Chemical and Environmental Engineering, Yale University, USA
6) Michael Baym - Assistant professor of biomedical informatics
Department of Biomedical Informatics, Harvard Medical School, USA
Referenties:
[1] Elbe, S., and Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges, 1:33-46, 2017. DOI:10.1002/gch2.1018
[2] Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). doi: 10.1038/nbt.3519
[3] Baaijens, J. A., Zulli, A., Ott, I. M., ..., Grubaugh, N. D., Peccia, J. and Baym, M. Variant abundance estimation for SARS-CoV-2 in wastewater using RNA-Seq quantification. medRxiv 2021.08.31.21262938; doi:https://doi.org/10.1101/2021.08.31.21262938
Terug naar overzicht